Е.Д. Білоусова, М.Ю. Никанорова, Е.А. Миколаєва
Московський НДІ педіатрії та дитячої хірургії МОЗ РФ
Стаття присвячена актуальній і маловідомою широкому колу лікарів проблеми - діагностиці та лікуванню вроджених дефектів метаболізму в неонатальному періоді. Описуються симптоми, на підставі яких педіатр і невролог можуть запідозрити у новонародженого метаболічних захворювань, наводиться алгоритм необхідних лабораторних досліджень. Коротко викладені загальні принципи лікування неонатальних вроджених дефектів метаболізму.
Діагностика та лікування вроджених аномалій метаболізму - одна з найскладніших завдань як для невролога, так і для педіатра. З одного боку, труднощі діагностики пов'язані з клінічним поліморфізмом захворювань, по-різному протікають у дітей різного віку, з іншого - з тим, що самі різні порушення обміну в одній і тій же віковій групі можуть мати подібні клінічні прояви [1].
Особливі труднощі викликає діагностика вроджених дефектів метаболізму у новонародженого [2]. Захворювання протікають важко і закінчуються летально до того, як стає очевидною специфічна метаболічна і неврологічна картина. У періоді новонародженості проявляються приблизно 25% відомих спадкових хвороб обміну речовин і майже всі вони відрізняються особливою тяжкістю стану дитини. За гостротою початку і перебігу спадкові хвороби обміну речовин в неонатальному періоді можуть нагадувати нейроінфекції і нейродістресс-синдром. Однак явна прогредиентность течії з втратою функцій не характерна для новонародженого, так як самі ці функції ще не сформовані. Тому більшість дітей з неонатальними формами спадкових дефектів метаболізму або помирають протягом перших місяців життя без встановленого діагнозу, або тривалий час спостерігаються з різними неадекватними діагнозами. Найчастіше спадкові хвороби обміну ховаються під маскою дитячого церебрального паралічу, розумової відсталості і епілепсії, резистентної до антиконвульсанти. Причини неонатальних спадкових метаболічних порушень численні (див. Нижче).
Етіологія спадкових захворювань обміну речовин з маніфестацією клінічних проявів в неонатальному періоді
Порушення метаболізму амінокислот
Порушення циклу синтезу сечовини (дефіцит N-ацетілглутаматсінтетази, карбамоілфосфатсінтетази, орнітінкарбамоілтрансферази, аргінінсукцінатсінтетази, аргінінсукцінатліази і аргінази)
Гіперорнітінемія - гипераммониемия - гомоцітруллінурія
Хвороба з запахом сечі кленового сиропу (лейциноз)
Порушення метаболізму органічних кислот
Некетотіческая гіпергліцінемію
изовалериановая ацидемія
пропіонова ацидемія
Метилмалонова ацидемія
мевалонової ацидурия
3-гідрокси-3-метілглутаровая ацидемія
мітохондріальні енцефалопатії
Порушення в системі піруватдегідрогеназного комплексу (дефіцит піруваткарбоксілази, піруватдегідрогенази, дігідроліпоілтрансацетілази і дігідроліпоілдегідрогенази) порушення функцій дихального ланцюга (дефіцити ферментів I-IV комплексу дихального ланцюга) порушення функцій ферментів циклу Кребса (дефіцит фумарази, сукцинатдегідрогенази, $ \ alpha $ -кетоглутаратдегідрогенази і аконітаза) порушення синтезу і обміну карнітину (системна недостатність карнітину, дефіцит карнітінпальмітоілтрансферази II і ацілкарнітінтранслокази) Порушення $ \ beta $ -Окислення жирних кислот (дефіцити ацил-СоА-дегідрогенази жирних кислот з короткою, середньої, довгою і дуже довгою вуглецевої ланцюгом, множинний дефіцит ацил-СоА-дегідрогенази)
Порушення обміну біотину
дефіцит біотинідази
Дефіцит синтетази голокарбоксілаз
хвороби пероксисом
синдром Цельвегера
неонатальна адренолейкодистрофія
Гіперпіпеколовая ацидемія
Дефіцит ацил-СоА-оксидази пероксисом
Дефіцит 3-оксіаціл-СоА-тіолази пероксисом
Біфункціонального недостатність протеїнів
лейкодистрофії
хвороба Краббе
Лейкодістрофія Канавана - Ван Богарта - Бертранд
метахроматіческая лейкодистрофия
Лейкодістрофія Пеліцеус - Мерцбахера
Суданофільние лейкодистрофии
Порушення обміну вуглеводів
Дефіцит фруктозо-1,6-альдолази
Спадковою непереносимістю фруктози
Галактоземия (дефіцит галактозо-1-фосфатуріділтрансферази)
Рідко, але зустрічаються неонатальні форми таких захворювань, як хвороба Менкеса, глікогеноз типу II, хвороба Німана-Піка і Гоше, синдром Барта, тирозинемия I типу, хвороба Альперса, а також сіалідоз [3].
Епідеміологія. Дані про поширеність найбільш вивчених спадкових метаболічних захворювань наведені в табл. 1.
Таблиця 1. Поширеність спадкових захворювань обміну речовин з неонатальним дебютом Захворювання Частота Дефіцит аргінінсукцінатліази 1/70 000 Дефіцит карбамоілфосфатсінтетази 1/1 000 000-1 / 800 000 Дефіцит аргінінсукцінатсінтетази 1/250 000 Некетотіческая гіпергліцінемію 1/55 000 Хвороба з запахом сечі кленового сиропу 1 / 200 000 Изовалериановая ацидемія 1/200 000 Пропіонова ацидемія 1/350 000 Метилмалонова ацидемія 1/48 000 Дефіцит ацил-СоА-дегідрогенази жирних кислот з середньою довжиною вуглецевого ланцюга 1/9 000-1 / 13 000 Синдром Цельвегера 1/50 000- 1/100 000 Дефіцит биот нідази 1/35 000-1 / 54 000 Дефіцит орнітінкарбамоілтрансферази 1/80 000 Дефіцит аргінінсукцінатліази 1/90 000 Тирозинемия I типу 1/30 000 Аргінінемія 1/990 000
Поширеність інших, менш вивчених дефектів метаболізму невідома. Число описів хворих з рідкісними неонатальними метаболічними захворюваннями коливається від одиничних до сотень спостережень.
Генетичні дані. Більшість вроджених дефектів метаболізму з проявами в неонатальному періоді мають аутосомно-рецесивний тип спадкування. Винятками з правила є неонатальна адренолейкодистрофія, лейкодистрофия Пеліцеус-Мерцбахера, хвороба Менкеса і синдром Барта, які успадковуються по рецесивним зчеплення з Х-хромосомою типом, і дефіцит орнітінкарбамоілтрансферази, який передається домінантно, в зчепленні з Х-хромосомою. Дефіцит піруватдегідрогенази і множинний дефіцит ацил-СоА-дегідрогенази мають два можливих типу успадкування - аутосомно-рецесивний і рецесивний, зчеплений з Х-хромосомою. Порушення в системі транспортної ланцюга електронів в дихальної ланцюга успадковуються по мітохондріального типу (материнське успадкування). Дефіцит I комплексу дихального ланцюга (NADH; CoQ-редуктази) успадковується як по аутосомно-рецесивним, так і по мітохондріального типу, описані також X-Cцепление форми.
Значних успіхів досягнуто в молекулярної діагностики неонатальних дефектів метаболізму. У табл. 2 представлені вроджені метаболічні захворювання з картірован генами.
Загальна клінічна характеристика вроджених дефектів метаболізму в неонатальному періоді
Незважаючи на те, що точний діагноз можливий тільки при застосуванні лабораторних методів дослідження, важливе значення мають анамнестичні та клінічні дані, на підставі яких лікар може запідозрити наявність у новонародженого вродженого дефекту метаболізму.
Анамнез. На спадкове захворювання метаболізму вказують відомості про можливе близькоспорідненому шлюбі батьків, неврологічна симптоматика у одного з батьків, наявність сібса, який страждає неврологічним захворюванням або померлого з неясної причини в неонатальному періоді, дитинстві або дитинстві. Як правило, тяжкість стану самого новонародженого не відповідає щодо благополучному перебігу даної вагітності і пологів, т. Е. Відсутні вказівки в анамнезі на важку гіпоксію плода і асфіксію новонародженого, травматичні ушкодження нервової системи.
Початок захворювання. Для багатьох неонатальних нейрометаболіческіх захворювань характерний <світлий проміжок> тривалістю в кілька днів від народження до перших клінічних проявів захворювання. <Світлий проміжок> є хоч і частим, але не обов'язковим симптомом при спадкових хворобах обміну речовин.
Порушення режиму сну - неспання. Для здорового доношеного новонародженого характерні періоди спонтанно виникає неспання, коли дитина прокидається, рухається і плаче. Для недоношеної з терміном гестації від 30 до 34 тижнів характерно в основному стимульоване неспання (при стимуляції дитина прокидається, рухається і плаче), є також короткочасне спонтанне неспання. У новонародженого з спадковим захворюванням обміну речовин можливі всі ступені тяжкості порушення рівня неспання (порушення свідомості) - від легкої сонливості до важкої коми.
Симптомокомплекс <млявого дитини> є типовим для спадкових аномалій метаболізму. Цим терміном позначається генералізована м'язова гіпотонія. Симптомокомплекс <млявого дитини> включає позу <жаби>, зниження опору до пасивним рухам, збільшення обсягу рухів в суглобах, зниження загальної рухової активності і затримку моторного розвитку. М'язова гіпотонія і зниження сухожильних рефлексів, характерні для спадкових дефектів метаболізму, пізніше (на 1-2-му році життя) змінюються м'язової дистонією і спастичністю. Винятком є неонатальна форма лейкодистрофии Краббе і хвороба з запахом сечі кленового сиропу, при яких вже в неонатальному періоді можливе підвищення м'язового тонусу.
Неонатальні судоми - один з найбільш частих симптомів вроджених дефектів метаболізму. Найбільш часто - це миоклонии і мінімальні судомні прояви (мимовільні рухи в області обличчя або кінцівок: насильницьке відкривання очей або моргання, тоническая девіація очних яблук, жувальні і ковтальні руху, смоктання і ін.). Як правило, судоми метаболічного генезу повторюються і погано піддаються лікуванню антиконвульсантами. Можливий епілептичний статус.
Незрозумілі труднощі вигодовування. Для спадкових дефектів метаболізму характерні порушення смоктання і ковтання. У важких випадках дитина не смокче груди і не ковтає, що вимагає зондового годування. У менш важких випадках смоктання порушено, новонароджений нещільно охоплює губами сосок, відзначається постійне підтікання молока з кутів рота і поперхіванія при ковтанні. Порушення смоктання і ковтання при вроджених дефектах обміну можуть мати різне походження: дисплазія рухових зон кори головного мозку, що відповідають за орофарінгіальную мускулатуру, за рахунок пренатального впливу токсичних метаболітів у плода, летаргія у новонародженого, загальна м'язова гіпотонія, яка захоплює і м'язи, що відповідають за смоктання і ковтання. Надалі у хворих відзначаються порушення жування і дизартрія, так як дані функції забезпечуються тієї ж мускулатурою.
Дихальні порушення нерідко зустрічаються при спадкових захворюваннях обміну речовин. Гіперпное і апное спостерігаються при відсутності захворювань серця та легень і респіраторного дистрес-синдрому. Їх виникнення пов'язане з впливом токсичних продуктів метаболізму на дихальний центр або супутнім метаболічним ацидозом.
Збільшення розмірів печінки і селезінки в поєднанні з неврологічною симптоматикою у новонародженого з високою ймовірністю свідчить про можливу наявність спадкового захворювання обміну речовин, хоча також може відзначатися при інфекційних і гематологічних хворобах. Збільшення розмірів печінки характерно для хвороб пероксисом, мітохондріальних енцефаломіопатія, порушень обміну амінокислот і органічних кислот, а також для більш рідкісних неонатальних форм хвороб накопичення (лейкодистрофии Краббе, хвороб Нимана - Піка і Гоше, сіалідоза і ін.).
Анорексія і блювота також відносяться до загальних симптомів, що спостерігається при багатьох вроджених метаболічних захворюваннях. Вони особливо характерні для гіперамоніємія, органічних ацидемій і порушень -Окислення жирних кислот. Більш рідкісним симптомом ураження шлунково-кишкового тракту є діарея. Як правило, всі ці симптоми викликають затримку збільшення маси у новонародженого, і їх легко прийняти за шлунково-кишкові захворювання.
Крім загальних клінічних проявів, характерним симптомом багатьох метаболічних захворювань є черепно-лицьова дисморфия (табл. 3).
Таблиця 3. Черепно-лицева дисморфия при різних спадкових захворюваннях обміну речовин з неонатальним дебютом Захворювання Черепно-лицева дисморфия Синдром Цельвегера і інші хвороби пероксисом Брахіцефалія, одутле, пласке обличчя з опухлими століттями, високе чоло, згладжені надбрівні дуги, гипертелоризм, епікант, монголоїдної розріз очей, микрогнатия * Множинний дефіцит ацил-СоА-дегідрогенази Високе чоло, низько розташовані вушні раковини, гипертелоризм дефіцит піруватдегідрогенази Кирпатий ніс з широкою спинкою, ні до посаджені ротирована вкінці вушні раковини, микрогнатия Дефіцит кофактора молібдену Трикутне обличчя, кирпатий ніс з широким переніссям, енофтальм, телекант, щілина м'якого піднебіння мевалонової ацидурия Широка перенісся, микрогнатия, низько розташовані вушні раковини * При інших хворобах пероксисом (крім хвороби Цельвегера) черепно- лицьова дисморфия менш постійна і менш виражена.
Характерні для окремих спадкових захворювань обміну речовин зміни шкіри, внутрішніх органів і очей наведені в табл. 4.
Таблиця 4. Екстраневрологіческіе прояви спадкових вроджених дефектів метаболізму в неонатальному періоді Об'єкт ураження Характер ураження Захворювання Kожа Еритематозні висипання Пропіонова ацідеміяМетілмалоновая ацидемія
Дефіцит синтетази голокарбоксілаз
Хвороба з запахом сечі кленового сиропу
Цітруллінемія
Аргінінянтарная ацидурия
Тирозинемия I типу
Дефіцит ацил-СоА-дегідрогенази жирних кислот з коротким вуглецевим ланцюгом
Очі Kатаракта Синдром Цельвегера і інші хвороби пероксісомГалактоземія
изовалериановая ацидемія
мевалонової ацидурия
Пігментна дегенерація сітківки Синдром Цельвегера і інші хвороби пероксісомДефіціт біотинідази Дисплазія сітківки Дефіцит сульфітоксідази і кофактора молібденаДефіціт біотинідази Дислокація кришталика. Глаукома Синдром Цельвегера і інші хвороби пероксисом Серце Kардіоміопатія Дефіцит I і IV комплексів дихальної цепіНарушеніе 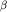 -Окислення мітохондріальних жирних кислот
-Окислення мітохондріальних жирних кислот
Дефіцит карнітінпальмітоілтрансферази II
3-гідрокси-3-метілглутаровая ацидемія
синдром Цельвегера
синдром Барта
Нирки Полікістоз Синдром Цельвегера і інші хвороби пероксісомДефіціт карнітінпальмітоілтрансферази II
Множинний дефіцит ацил-СоА-дегідрогенази
Полікістоз + диспластические зміни. Синдром Де Тоні - Дебре - Фанконі Дефіцит III і IV комплексів дихального ланцюга Підшлункова залоза Панкреатит Изовалериановая ацідеміяМетілмалоновая ацидурия
3-гідрокси-3-метілглутаровая ацидемія
Хвороба з запахом сечі кленового сиропу
Специфічний запах сечі є важливою діагностичною ознакою і зустрічається при лейциноз (запах <кленового сиропу> або сіна), ізовалеріанової ацидемії і множині дефіциті ацил-СоА-дегідрогенази (і при тому, і при іншому захворюванні характерний запах <сиру »або« спітнілих ніг> ), дефиците биотинидазы, дефиците синтетазы голокарбоксилаз, -метилкротонилглицинурии (запах <кошачей мочи>), тирозинемии I типа (запах <кипящей капусты>) и при болезни с запахом мочи кленового сиропа [5].
Клінічна діагностика. У типовому випадку спадкового дефекту метаболізму новонароджений народжується доношеною від фізіологічно протікають пологів, і його стан після народження цілком благополучно. Через деякий час (1-3 дні) стан дитини погіршується, розвиваються летаргія або ступор, з'являються судоми, дихальні розлади, анорексія і блювота. У зарубіжній літературі такий стан отримало найменування <нейродістресс>. Для неврологічного статусу характерні дифузна м'язова гіпотонія і порушення смоктання і ковтання. Підозра на наявність вродженого дефекту метаболізму отримує підтвердження, якщо є обтяженість родоводу по неврологічним захворюванням, специфічний запах сечі і екстраневрологіческіе прояви - кардіоміопатія, гепатоспленомегалія і ін.
Лабораторна діагностика спадкових дефектів метаболізму в неонатальному періоді
При підозрі на спадкове порушення обміну речовин рекомендується на першому етапі проведення стандартних клінічних та біохімічних досліджень крові. Про можливу наявність метаболічного захворювання свідчать:
- анемія, лейко- і тромбоцитопенія (характерні для ізовалеріанової, пропіонової і метилмалоновой ацидемій);
- нейтропенія (характерна для синдрому Барта);
- ацидоз у поєднанні з кетозом (характерний для більшості органічних ацидемій);
- підвищення вмісту лактату в крові (характерно для порушень метаболізму пірувату, дефектів дихальних комплексів і ферментів -Окислення жирних кислот);
- гіпоглікемія з низьким вмістом кетонових тіл (характерна для порушень мітохондріального -Окислення жирних кислот);
- гипераммониемия (характерна для порушень циклу сечовини, дефіциту ацил-СоА-дегідрогенази жирних кислот з середньою довжиною вуглецевого ланцюга і для більшості органічних ацидемій).
На другому етапі обстеження при наявності хоча б одного з перерахованих вище лабораторних порушень показано більш детальне лабораторне обстеження новонародженого, що включає визначення спектра амінокислот крові та сечі, органічних кислот в сечі, карнітину і ацилкарнітину в крові (див. Таб. 5).
Наступним етапом діагностики є визначення активності ферментів, дефіцитом яких викликано захворювання, в культурі фібробластів, лімфоцитах або биоптатах м'язи і печінки. На завершальному етапі проводиться молекулярна діагностика патології за допомогою ДНК-зондів.
Нейросонографічного дослідження в неонатальному періоді, як правило, малоспеціфічни і більш важливі для диференціального діагнозу з гіпоксично-ішемічними ураженнями і внутрішньочерепними крововиливами у новонароджених [6]. Можлива наявність пренатальних поразок кори і білої речовини, базальних ядер і мозолистого тіла у вигляді зниження щільності зазначених утворень. Більш інформативною є магнітно-резонансна томографія головного мозку, яка проводиться на 1-му році життя дитини і може демонструвати кортикальні дисгенезії, гіпоплазію мозолистого тіла і мозочка, а також пошкодження білої речовини головного мозку.
Результати електроенцефалографії, електронейроміографія, дослідження викликаних коркових потенціалів в неонатальному періоді неспецифічні і не можуть використовуватися для диференціальної діагностики.
Диференціальний діагноз. Подібні зі спадковими дефектами метаболізму клінічні симптоми (неврологічні симптоми, порушення дихання і свідомості, судоми, м'язова гіпотонія, блювання) часто зустрічаються при гіпоксично-ішемічних енцефалопатіях, пороках розвитку головного мозку, внутрішньоутробних інфекціях або внутрішньочерепних крововиливах. Основні анамнестические і клінічні відмінності спадкових хвороб обміну і гіпоксично-ішемічної енцефалопатії новонароджених наведені в табл. 6.
Таблиця 6. Диференціальний діагноз спадкових захворювань обміну речовин і гіпоксично-ішемічної енцефалопатії в неонатальному періоді Симптом Гипоксически-ішемічна енцефалопатія Спадкова хвороба метаболізму Обтяжливість родоводу по неврологічним захворюванням - + Гіпоксія плода + - обтяження перебігу вагітності та пологів + - Захворювання матері, що викликають порушення плацентарного кровотоку (хронічні запальні захворювання статевої сфери, патологія плаценти, артеріальна гіпертензія та ін.) + - меконіальної фарбування навколоплідних вод + - Недоношенность + * Поява неврологічних порушень відразу після народження + - Черепно-лицева дисморфия - + Гепатомегалия, кардіоміопатія, полікістоз нирок - + нейросонографічного зміни, характерні для гіпоксично-ішемічної енцефалопатії + - Примітка. * Можлива, але не характерна.
Лікування. У багатьох випадках необхідні екстрені терапевтичні заходи, спрямовані на боротьбу з жізнеугрожающімі ацидозом, гіпоглікемією, гіперамоніємією і гіповолемією [7]. Концентрація токсичних метаболітів в крові повинна бути нормалізована якомога швидше, щоб запобігти необоротне ушкодження головного мозку. Найбільш ефективним способом лікування в гострому періоді є гемодіаліз.
Принципи лікування генетично детермінованих дефектів метаболізму можна умовно розділити на <прямі "і" непрямі> [2]. До <прямим> підходам відносять заміну дефектного ферменту або гена, відповідального за синтез ферменту. Даний підхід забезпечує кардинальне рішення проблем, пов'язаних з метаболічними розладами. Однак, на жаль, <пряме> втручання в даний час неможливо.
До <непрямим> підходам до терапії відносяться:
- зменшення надходження в організм речовин, обмін яких порушений;
- введення відсутнього метаболіти;
- видалення токсичних речовин;
- зведення до мінімуму введення препаратів або дії інших факторів, що сприяють погіршенню стану хворого;
- призначення вітамінів, які є кофакторами дефектних ферментів;
- зменшення утворення токсичних метаболітів.
Зменшення введення погано метаболізуються речовин досягається за допомогою модифікацій дієти. Даний метод використовується у хворих з хворобами обміну амінокислот, порушеннями в циклі синтезу сечовини, при деяких органічних ацидурія і мітохондріальних енцефаломіопатія. У лікуванні порушень окислення жирних кислот застосовується дієтотерапія з обмеженням жирів (до 15% калорійності) і збагаченням вуглеводами (до 65% калорійності).
Зв'язування токсичного метаболіту іншими речовинами і виведення його з організму у вигляді нетоксичних сполук реалізується за допомогою застосування спеціальних лікарських засобів. Так, при гіперамоніємії використовуються бензоат, фенілацетат і фенілбутірат натрію. Бензоат натрію зв'язується з гліцином і у вигляді нетоксичного з'єднання виводиться з організму. При неефективності даних препаратів використовується гемо- або перитонеальний діаліз. Паралельно при гіперамоніємії проводиться різке обмеження в дієті білка і внутрішньовенно вводиться глюкоза. Використання даних методик лікування призвело до того, що 93% дітей з порушеннями циклу синтезу сечовини доживають до 1 року. При ізовалеріянової ацидемії з метою виведення токсичних метаболітів використовується гліцин, при дефектах -Окислення жирних кислот - карнітин каксредство, що підвищує інтенсивність внутрішньоклітинного транспорту жирних кислот.
До факторів, що провокує погіршення перебігу деяких дисметаболических захворювань (наприклад, дефіциту ацил-СоА-дегідрогенази) в неонатальному періоді та після нього, ставляться тривале голодування, надлишок білків і жирів в дієті і інтеркурентних захворювання. Нерідко вони <запускають> каскад незворотних метаболічних порушень. Тому при багатьох спадкових хворобах обміну речовин слід уникати білкових або жирових харчових навантажень або голодування, проводити лікування інтеркурентних хвороб.
Призначення високих доз вітамінів (тіаміну 200-1000 мг / сут, рибофлавіну 100 мг / cyт і нікотинаміду 1 г / сут) дає лікувальний ефект при різних видах мітохондріальних енцефаломіопатія. Деякі рібофлавінзавісімие форми множинного дефіциту ацил-СоА-дегідрогенази (глутаровая ацидемія, тип II) піддаються лікуванню високими дозами рибофлавіну (до 100 мг / добу).
Основні види терапії спадкових хвороб метаболізму з неонатальним дебютом представлені в табл. 7.
Таблиця 7. Особливості терапії при неонатальних формах спадкових захворювань обміну речовин Захворювання Лікування Порушення в циклі синтезу сечовини Гемодіаліз, перитонеальний діалізВисококалорійная дієта з різким обмеженням вмісту білка і добавками амінокислот
Внутрішньовенне або ентеральне введення бензоату натрію (250 мг / кг на добу) або фенілацетат (250-500 мг / кг на добу) або фенілбутірата (500 мг / кг на добу)
L-аргінін (400-800 мг / кг на добу)
пересадка печінки
Некетотіческая гіпергліцінемію Внутрішньовенне введення бензоату натрію (250 мг / кг на добу) Піридоксин (50 мг / кг на добу)
декстрометорфан
Протипоказаний прийом вальпроатов
Хвороба з запахом сечі кленового сиропу Гемодіаліз, перитонеальний діалізВисококалорійная дієта з обмеженням амінокислот з розгалуженим ланцюгом (40-60 мг лейцину на 1 кг маси)
Тіамін (50 мг - 500 мг / добу)
Изовалериановая ацидемія Дієта з обмеженням змісту белкаL-карнітин (100 мг / добу)
Гліцин (до 250 мг / кг / добу)
Метилмалонова ацидурия Дієта з різким обмеженням вмісту білка (ізолейцин, валін, метіонін, треонін) гідроксікобаламін, аденозилкобаламін (15 мг / добу)
L-карнітин (100 мг / кг на добу)
Метронідазол (10-15 мг / добу)
Пропіонова ацидемія Гемодіаліз, перитонеальний діалізДіета з різким обмеженням вмісту білка (ізолейцин, валін, метіонін, треонін)
L-карнітин (100 мг / кг на добу)
Біотин (5-10 мг / добу)
Метронідазол (10-15 мг / добу)
Дефіцит 3-гідро-3-метилглутарил-СоА-ліази Часте годування для виключення голоданіяДіета з низьким вмістом білка і ліпідів (вуглеводне харчування)
L-карнітин (100 мг / кг на добу)
Дефіцит фруктозо-1,6-біфосфатази Дієта з виключенням фруктозиВнутрівенное введення глюкози і бікарбонату Дефіцит піруватдегідрогеназного комплексу Дієта з високим вмістом жирів і низьким - углеводовL-карнітин (100 мг / кг на добу)
Тіамін (до 200 мг / добу)
Діхлорацетат (35-50 мг / кг на добу)
Ліпоєва кислота (до 500 мг / добу)
Дефіцит піруваткарбоксілази Дієта з високим вмістом жирів і низьким - углеводовБіотін (10 мг / добу)
Аспартатовая кислота
Дефіцит I комплексу дихального ланцюга мітохондрій Дієта з обмеженням углеводовНікотінамід (до 500 мг / добу)
Рибофлавін (до 100 мг / добу)
L-карнітин (50-100 мг / кг на добу)
Діхлорацетат (35-50 мг / кг на добу)
Множинна недостатність ацил-СоА-дегідрогенази жирних кислот Дієта з низьким вмістом білка і ліпідовРібофлавін (100-300 мг / добу)
L-карнітину (100 мг / кг на добу)
Дефіцит ацил-СоА-дегідрогенази жирних кислот з середньою довжиною вуглецевого ланцюга Часте годування для виключення голоданіяДіета зі зниженим вмістом жирів і підвищеним - вуглеводів
L-карнітин (100 мг / кг на добу)
Рибофлавін (100-300 мг / добу)
Внутрішньовенне введення розчинів глюкози і бікарбонату натрію
Значення своєчасної діагностики вроджених дефектів метаболізму надзвичайно велике. До доступним скринінговим методикам при спадкових хворобах обміну речовин з проявами в неонатальному періоді відносяться визначення вмісту лактату, глюкози і амонію в крові, органічних кислот і амінокислот в сечі. Несвоєчасне встановлення діагнозу і відсутність адекватного лікування може призвести до смерті новонародженого або до важкої неврологічної інвалідності. Правильне трактування патології здатна забезпечити оптимальне нервово-психічний розвиток дитини і надати членам його сім'ї своєчасну спеціалізовану медико-генетичну допомогу.
література
1. Вельтищев Ю.Є., Казанцева Л.З., Семячкіна А.Н. Спадкові хвороби обміну. Спадкова патологія людини. Під загальною редакцією Ю.Є. Вельтіщева, Н.П. Бочкова. М 1992; 41- 101.
2. The Neurology of Neonatal Hereditary Metabolic Diseases. Neurology of Hereditary Metabolic Diseases of Children. Ed. G.Lyon, RD Adams, EH Kolodny. New York: McGraw-Hill 1996; 6-44.
3. Спадкові хвороби і синдроми у дітей з порушеннями нервово-психічного розвитку. Під редакцією П.А. Темина, Л.З. Казанцевой. У пресі.
4. Вельтищев Ю.Є., Темін П.А., Білоусова Е.Д. Мітохондріальні хвороби, обумовлені мутаціями ядерної ДНК. Спадкові хвороби нервової системи. М: Медицина 1998; 409- 469.
5. Миколаєва Е.А. Органічні ацидемії, що супроводжуються судомним синдромом. Діагностика та лікування епілепсії у дітей. Можайськ: Терра 1997; 395-426.
6. Metabolic Diseases of the Nervous System. Textbook of Child Neurology. Ed. JH Menkes. Baltimore: Williams and Wilkins 1995; 29-151.
7. Metabolic and Degenerative Diseases of the Central Nervous System. Pathology, Biochemistry, and Genetics. Ed. J. Cervos-Navarro, H. Urich. San Diego: Academic Press 1995; 873.
А втори: Білоусова Є. Д., Никанорова М. Ю., Миколаєва Є. А.
Джерело: Російський вісник перинатології та педіатрії, 2000., N6, с.12-19
опубліковано 11/12/2009 16:39
оновлено 13/05/2011
- Вроджені і спадкові хвороби, генетика